
In an article lately revealed in Bodily Assessment Analysis, we present how deep studying might help resolve the basic equations of quantum mechanics for real-world programs. Not solely is that this an vital basic scientific query, but it surely additionally might result in sensible makes use of sooner or later, permitting researchers to prototype new supplies and chemical syntheses in silico earlier than making an attempt to make them within the lab. Immediately we’re additionally releasing the code from this examine in order that the computational physics and chemistry communities can construct on our work and apply it to a variety of issues. We’ve developed a brand new neural community structure, the Fermionic Neural Community or FermiNet, which is well-suited to modeling the quantum state of huge collections of electrons, the basic constructing blocks of chemical bonds. The FermiNet was the primary demonstration of deep studying for computing the power of atoms and molecules from first ideas that was correct sufficient to be helpful, and it stays essentially the most correct neural community methodology so far. We hope the instruments and concepts developed in our AI analysis at DeepMind might help resolve basic issues within the pure sciences, and the FermiNet joins our work on protein folding, glassy dynamics, lattice quantum chromodynamics and lots of different tasks in bringing that imaginative and prescient to life.
A Temporary Historical past of Quantum Mechanics
Point out “quantum mechanics” and also you usually tend to encourage confusion than the rest. The phrase conjures up photographs of Schrödinger’s cat, which might paradoxically be each alive and lifeless, and basic particles which can be additionally, one way or the other, waves. In quantum programs, a particle resembling an electron doesn’t have a precise location, as it will in a classical description. As a substitute, its place is described by a likelihood cloud – it’s smeared out all over the place it’s allowed to be. This counterintuitive state of affairs led Richard Feynman to declare: “If you happen to assume you perceive quantum mechanics, you don’t perceive quantum mechanics.” Regardless of this spooky weirdness, the meat of the idea might be decreased right down to only a few simple equations. Probably the most well-known of those, the Schrödinger equation, describes the conduct of particles on the quantum scale in the identical means that Newton’s legal guidelines describe the conduct of objects at our extra acquainted human scale. Whereas the interpretation of this equation could cause infinite head-scratching, the maths is far simpler to work with, resulting in the widespread exhortation from professors to “shut up and calculate” when pressed with thorny philosophical questions from college students.
These equations are ample to explain the conduct of all of the acquainted matter we see round us on the stage of atoms and nuclei. Their counterintuitive nature results in all types of unique phenomena: superconductors, superfluids, lasers and semiconductors are solely doable due to quantum results. However even the common-or-garden covalent bond – the fundamental constructing block of chemistry – is a consequence of the quantum interactions of electrons. As soon as these guidelines had been labored out within the Nineteen Twenties, scientists realised that, for the primary time, they’d an in depth idea of how chemistry works. In precept, they may simply arrange these equations for various molecules, resolve for the power of the system, and determine which molecules had been secure and which reactions would occur spontaneously. However once they sat down to truly calculate the options to those equations, they discovered that they may do it precisely for the only atom (hydrogen) and just about nothing else. All the pieces else was too sophisticated.
The heady optimism of these days was properly summed up by Paul Dirac:
The underlying bodily legal guidelines essential for the mathematical idea of a big a part of physics and the entire of chemistry are thus utterly identified, and the issue is simply that the precise software of those legal guidelines results in equations a lot too sophisticated to be soluble. It due to this fact turns into fascinating that approximate sensible strategies of making use of quantum mechanics must be developed
Paul Dirac, 1929
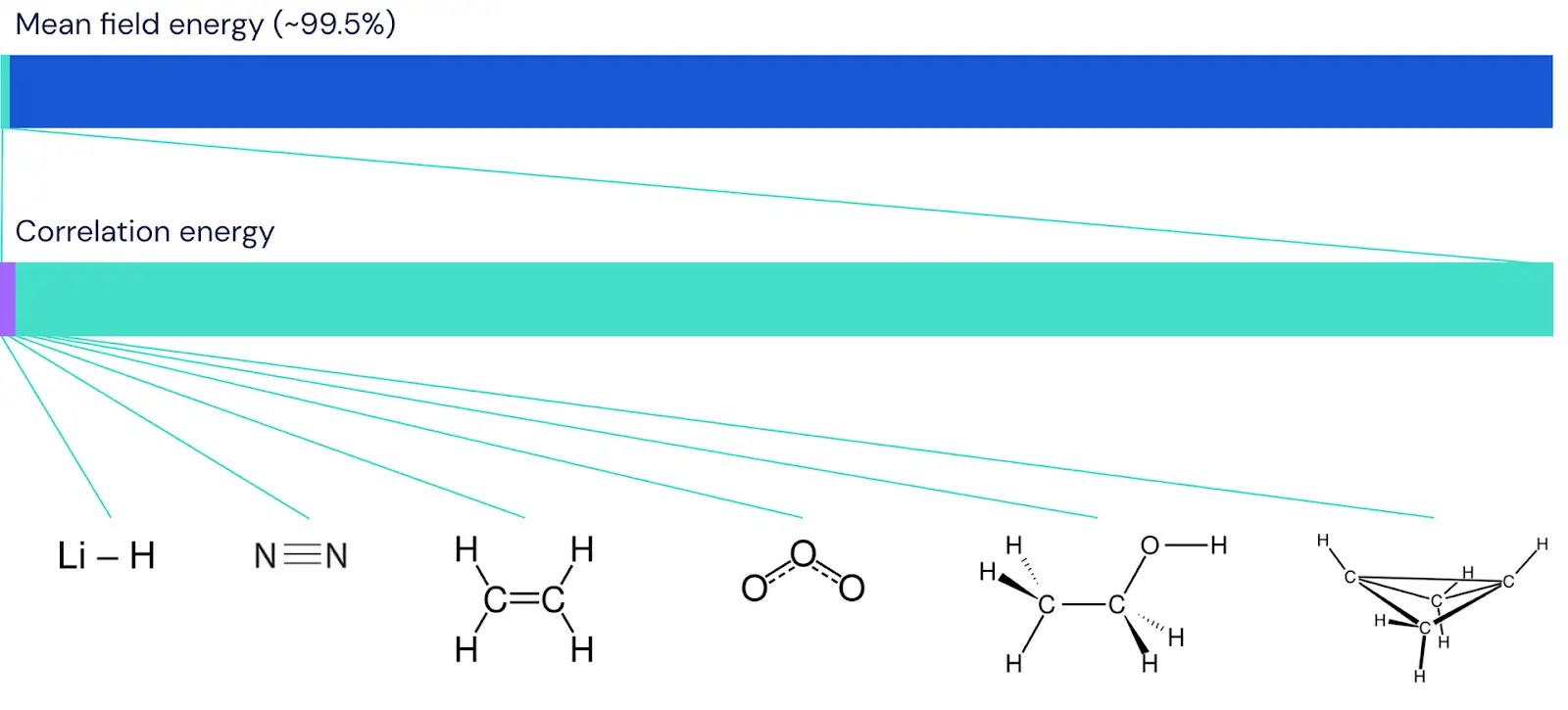
Many took up Dirac’s cost, and shortly physicists constructed mathematical methods that might approximate the qualitative conduct of molecular bonds and different chemical phenomena. These strategies began from an approximate description of how electrons behave that could be acquainted from introductory chemistry. On this description, every electron is assigned to a specific orbital, which supplies the likelihood of a single electron being discovered at any level close to an atomic nucleus. The form of every orbital then depends upon the typical form of all different orbitals. As this “imply area” description treats every electron as being assigned to only one orbital, it’s a very incomplete image of how electrons really behave. Nonetheless, it is sufficient to estimate the entire power of a molecule with solely about 0.5% error.
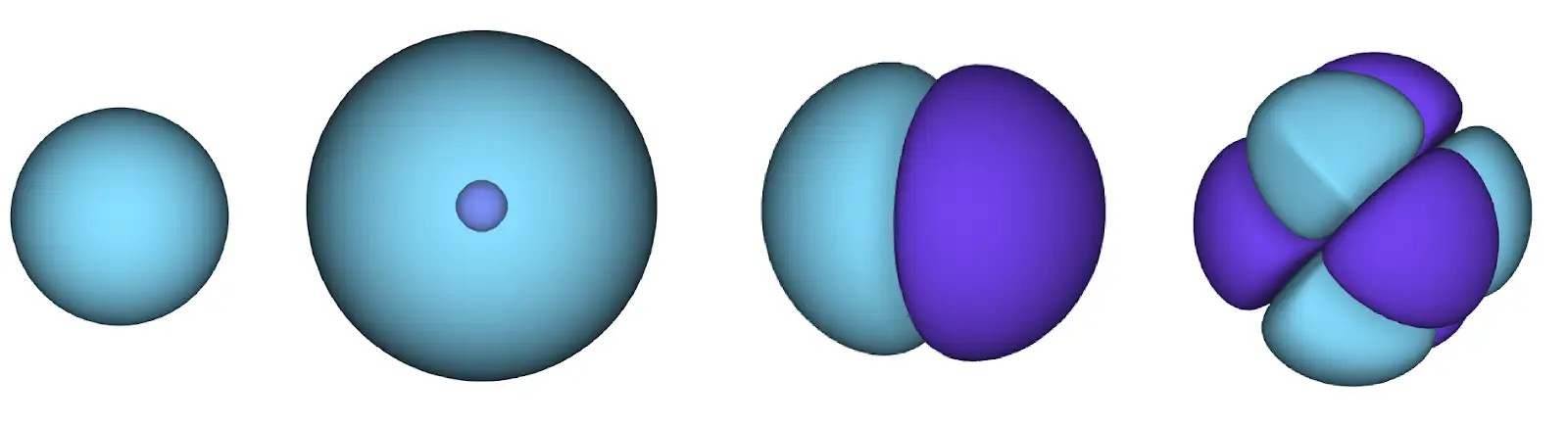
Sadly, 0.5% error nonetheless isn’t sufficient to be helpful to the working chemist. The power in molecular bonds is only a tiny fraction of the entire power of a system, and appropriately predicting whether or not a molecule is secure can typically depend upon simply 0.001% of the entire power of a system, or about 0.2% of the remaining “correlation” power. For example, whereas the entire power of the electrons in a butadiene molecule is sort of 100,000 kilocalories per mole, the distinction in power between completely different doable shapes of the molecule is simply 1 kilocalorie per mole. That signifies that if you wish to appropriately predict butadiene’s pure form, then the identical stage of precision is required as measuring the width of a soccer area right down to the millimeter.
With the arrival of digital computing after World Warfare II, scientists developed a complete menagerie of computational strategies that went past this imply area description of electrons. Whereas these strategies are available in a bewildering alphabet soup of abbreviations, all of them typically fall someplace on an axis that trades off accuracy with effectivity. At one excessive, there are strategies which can be primarily actual, however scale worse than exponentially with the variety of electrons, making them impractical for all however the smallest molecules. On the different excessive are strategies that scale linearly, however are usually not very correct. These computational strategies have had an unlimited impression on the apply of chemistry – the 1998 Nobel Prize in chemistry was awarded to the originators of many of those algorithms.
Fermionic Neural Networks
Regardless of the breadth of present computational quantum mechanical instruments, we felt a brand new methodology was wanted to deal with the issue of environment friendly illustration. There’s a purpose that the biggest quantum chemical calculations solely run into the tens of 1000’s of electrons for even essentially the most approximate strategies, whereas classical chemical calculation methods like molecular dynamics can deal with tens of millions of atoms. The state of a classical system might be described simply – we simply have to trace the place and momentum of every particle. Representing the state of a quantum system is much tougher. A likelihood must be assigned to each doable configuration of electron positions. That is encoded within the wavefunction, which assigns a optimistic or destructive quantity to each configuration of electrons, and the wavefunction squared provides the likelihood of discovering the system in that configuration. The house of all doable configurations is big – for those who tried to characterize it as a grid with 100 factors alongside every dimension, then the variety of doable electron configurations for the silicon atom could be bigger than the variety of atoms within the universe!
That is precisely the place we thought deep neural networks might assist. Within the final a number of years, there have been enormous advances in representing advanced, high-dimensional likelihood distributions with neural networks. We now know methods to practice these networks effectively and scalably. We surmised that, given these networks have already confirmed their mettle at becoming high-dimensional features in synthetic intelligence issues, perhaps they may very well be used to characterize quantum wavefunctions as properly. We weren’t the primary individuals to consider this – researchers resembling Giuseppe Carleo and Matthias Troyer and others have proven how fashionable deep studying may very well be used for fixing idealised quantum issues. We wished to make use of deep neural networks to deal with extra lifelike issues in chemistry and condensed matter physics, and that meant together with electrons in our calculations.
There is only one wrinkle when coping with electrons. Electrons should obey the Pauli exclusion precept, which signifies that they will’t be in the identical house on the identical time. It is because electrons are a sort of particle referred to as fermions, which embody the constructing blocks of most matter – protons, neutrons, quarks, neutrinos, and so forth. Their wavefunction have to be antisymmetric – for those who swap the place of two electrons, the wavefunction will get multiplied by -1. That signifies that if two electrons are on high of one another, the wavefunction (and the likelihood of that configuration) might be zero.
This meant we needed to develop a brand new kind of neural community that was antisymmetric with respect to its inputs, which we’ve got dubbed the Fermionic Neural Community, or FermiNet. In most quantum chemistry strategies, antisymmetry is launched utilizing a operate referred to as the determinant. The determinant of a matrix has the property that for those who swap two rows, the output will get multiplied by -1, similar to a wavefunction for fermions. So you may take a bunch of single-electron features, consider them for each electron in your system, and pack the entire outcomes into one matrix. The determinant of that matrix is then a correctly antisymmetric wavefunction. The most important limitation of this strategy is that the ensuing operate – referred to as a Slater determinant – will not be very basic. Wavefunctions of actual programs are normally much more sophisticated. The everyday means to enhance on that is to take a big linear mixture of Slater determinants – generally tens of millions or extra – and add some easy corrections based mostly on pairs of electrons. Even then, this is probably not sufficient to precisely compute energies.
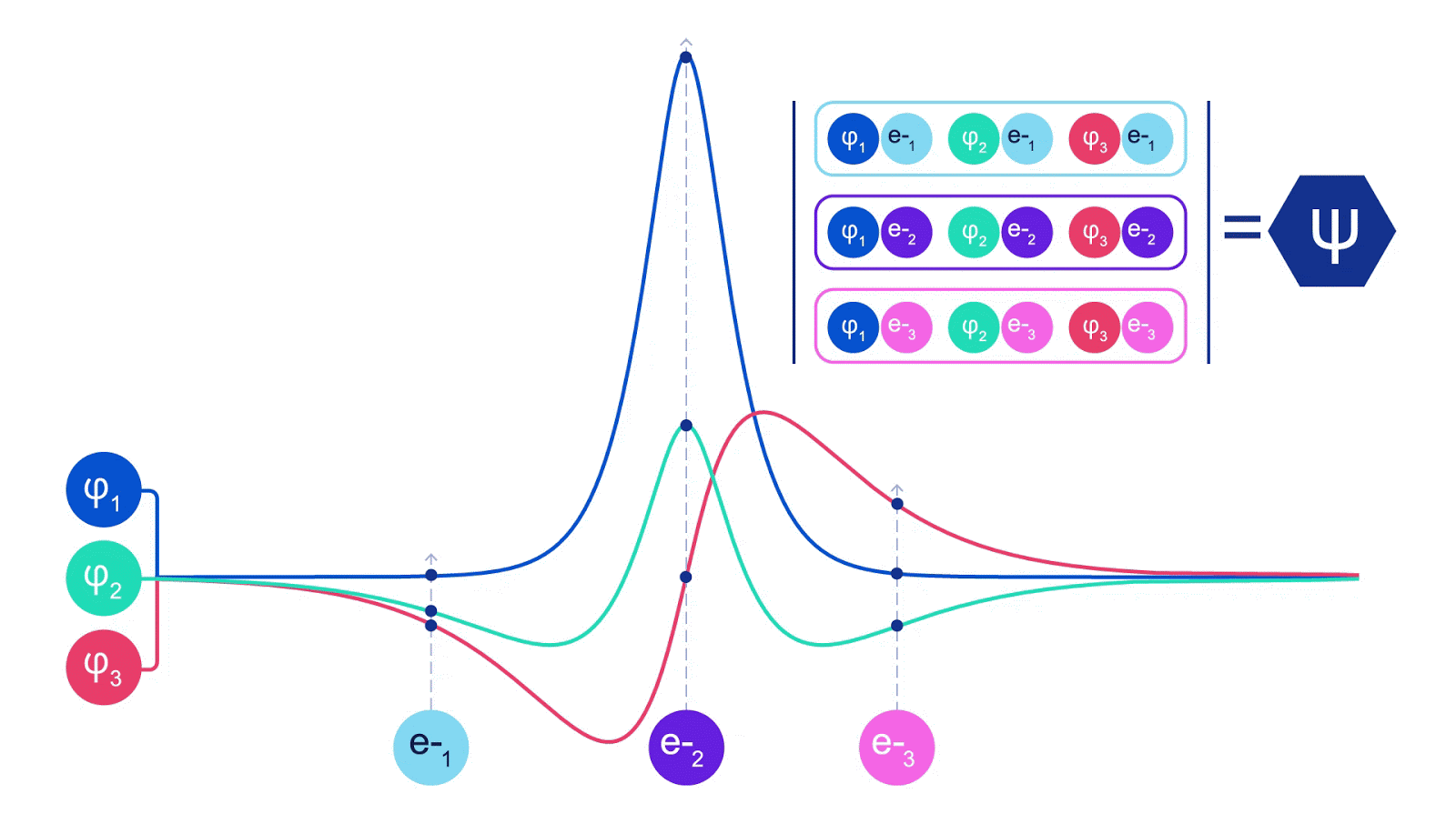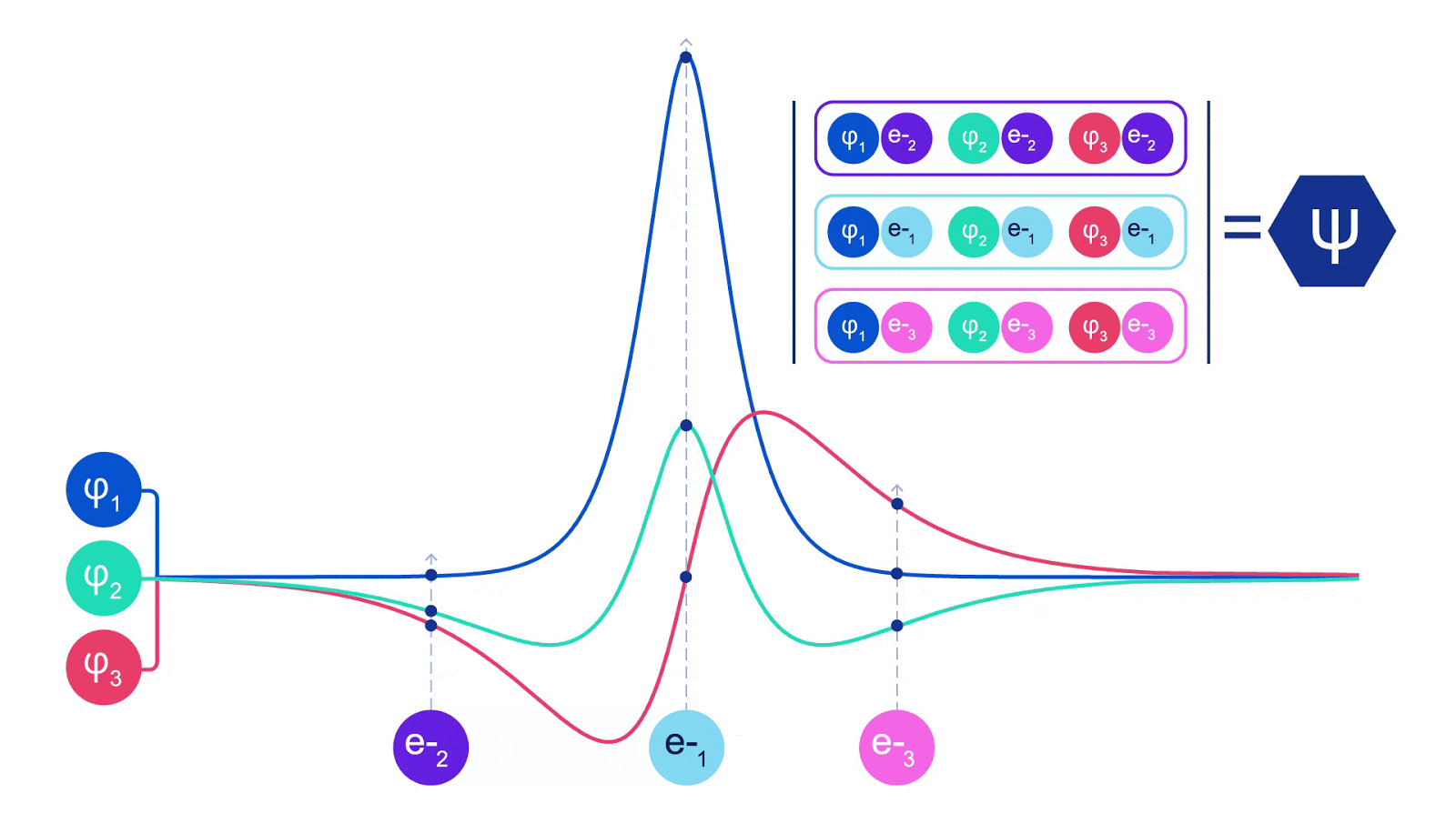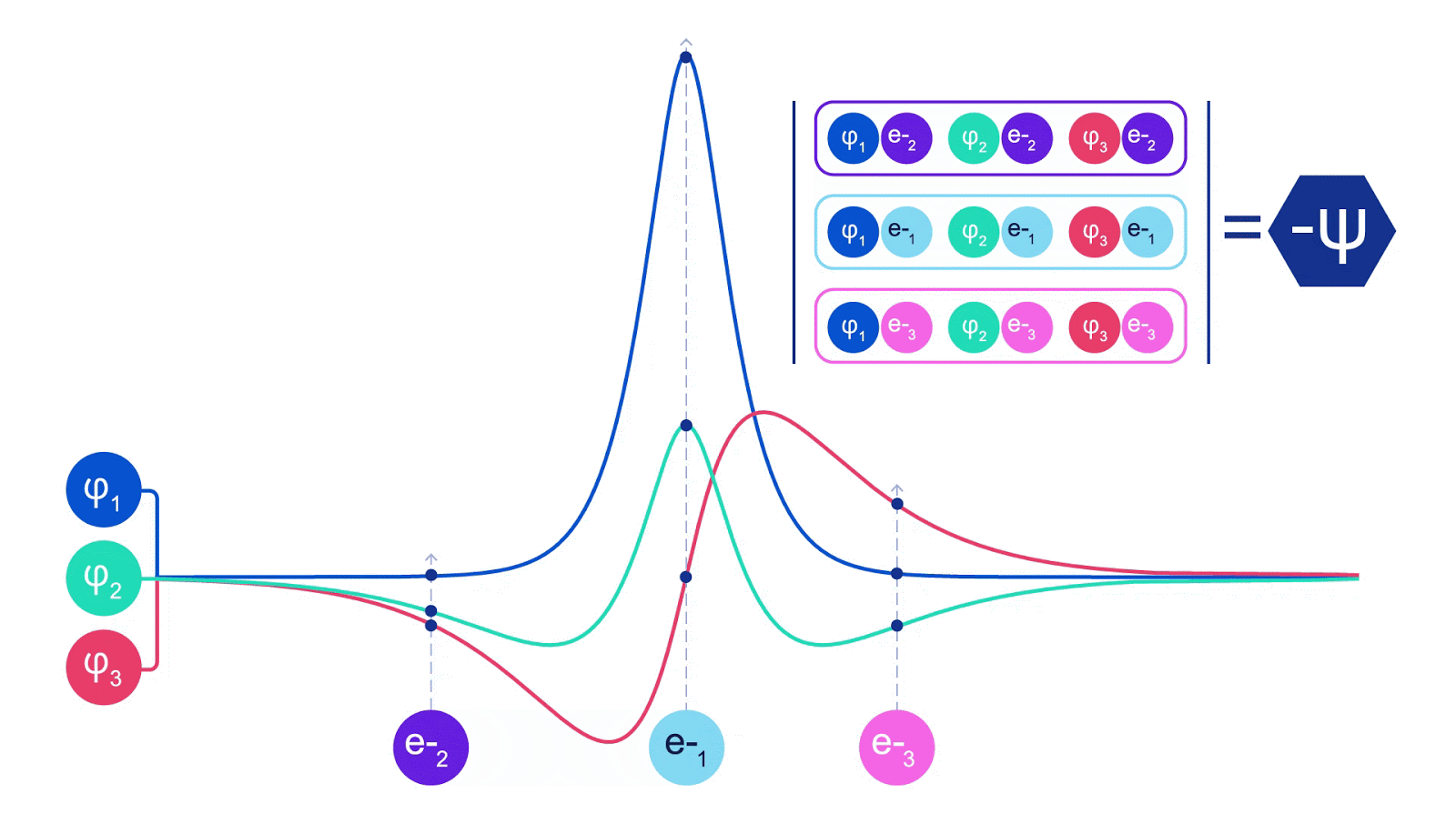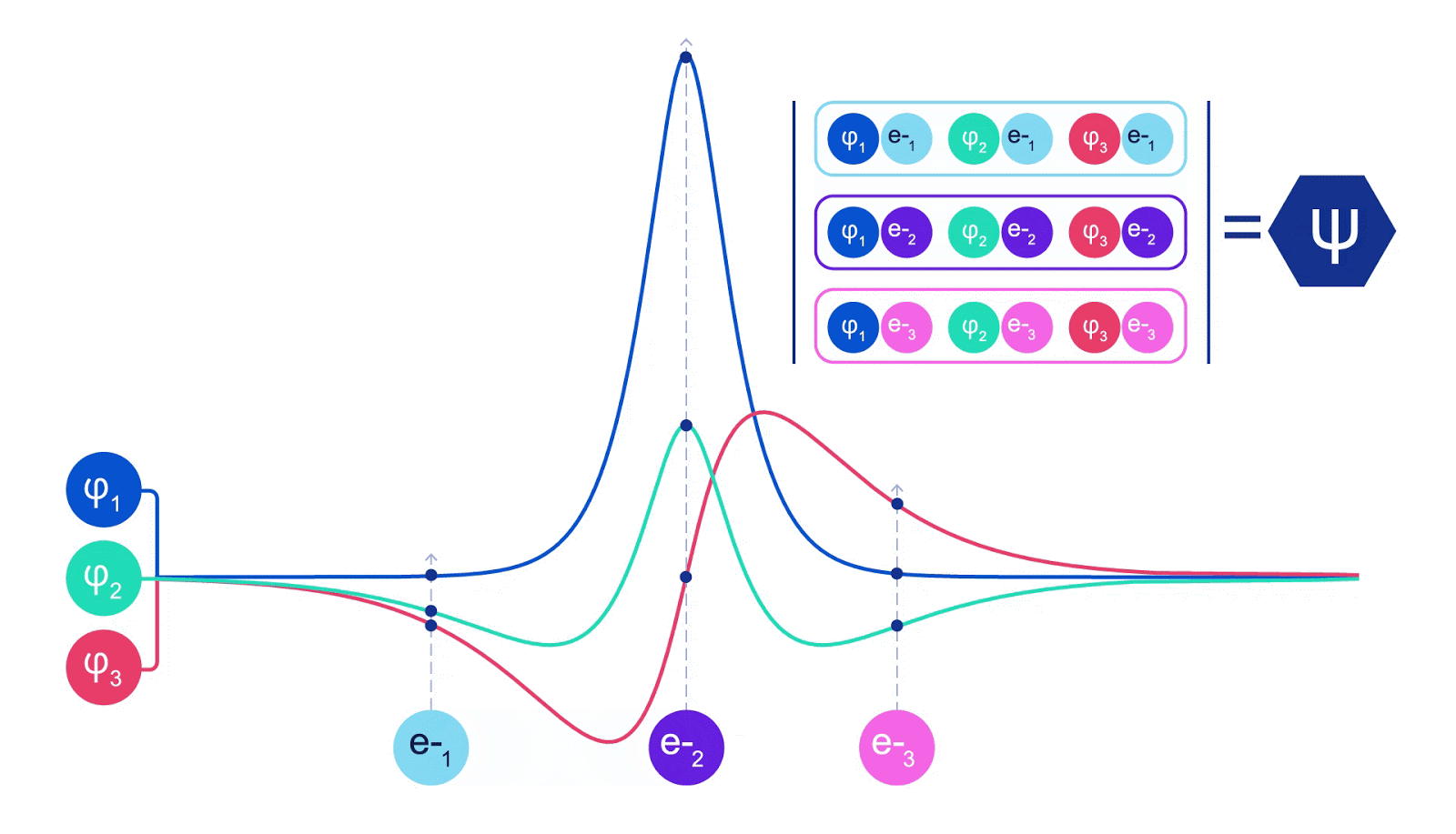
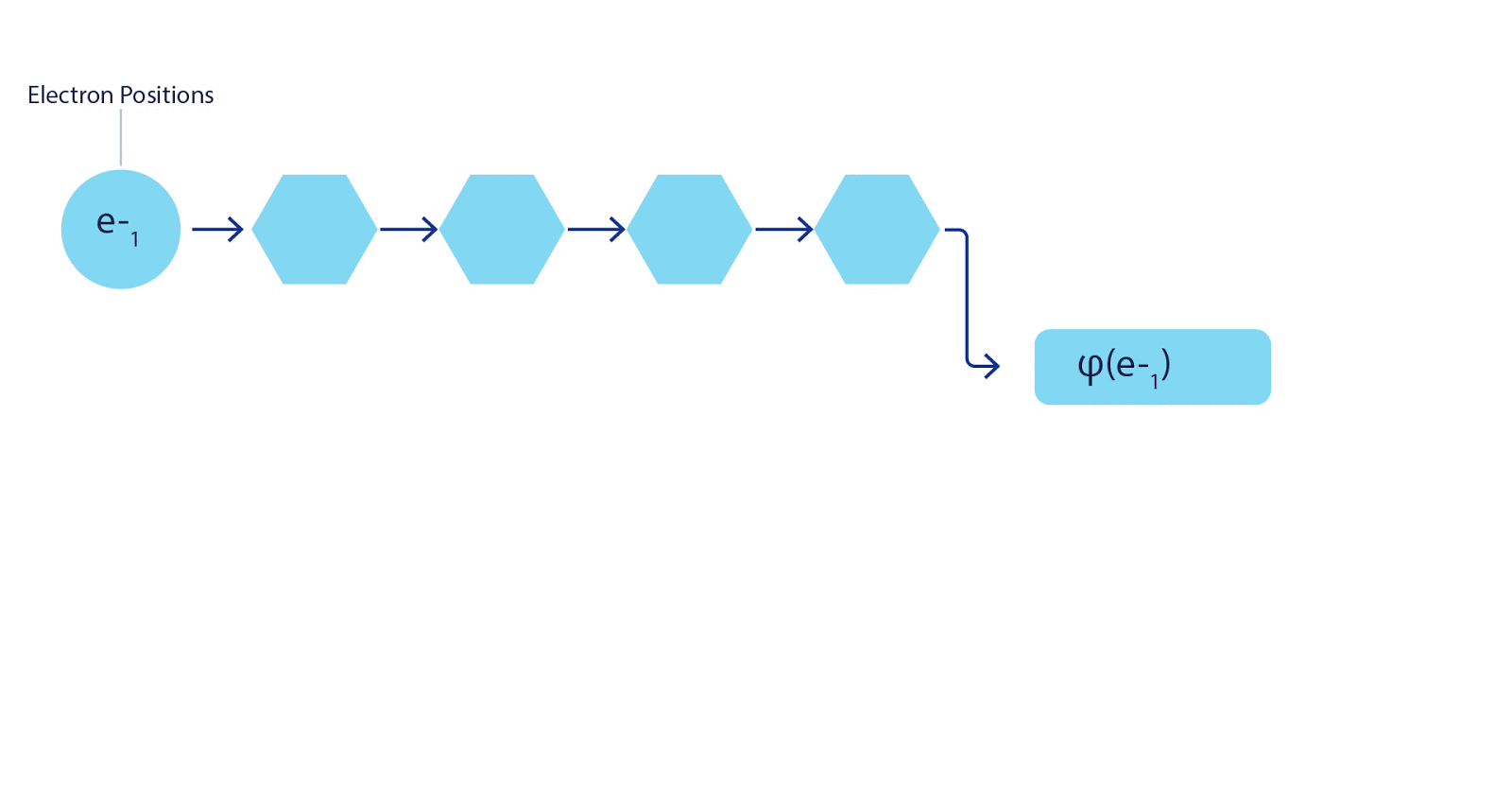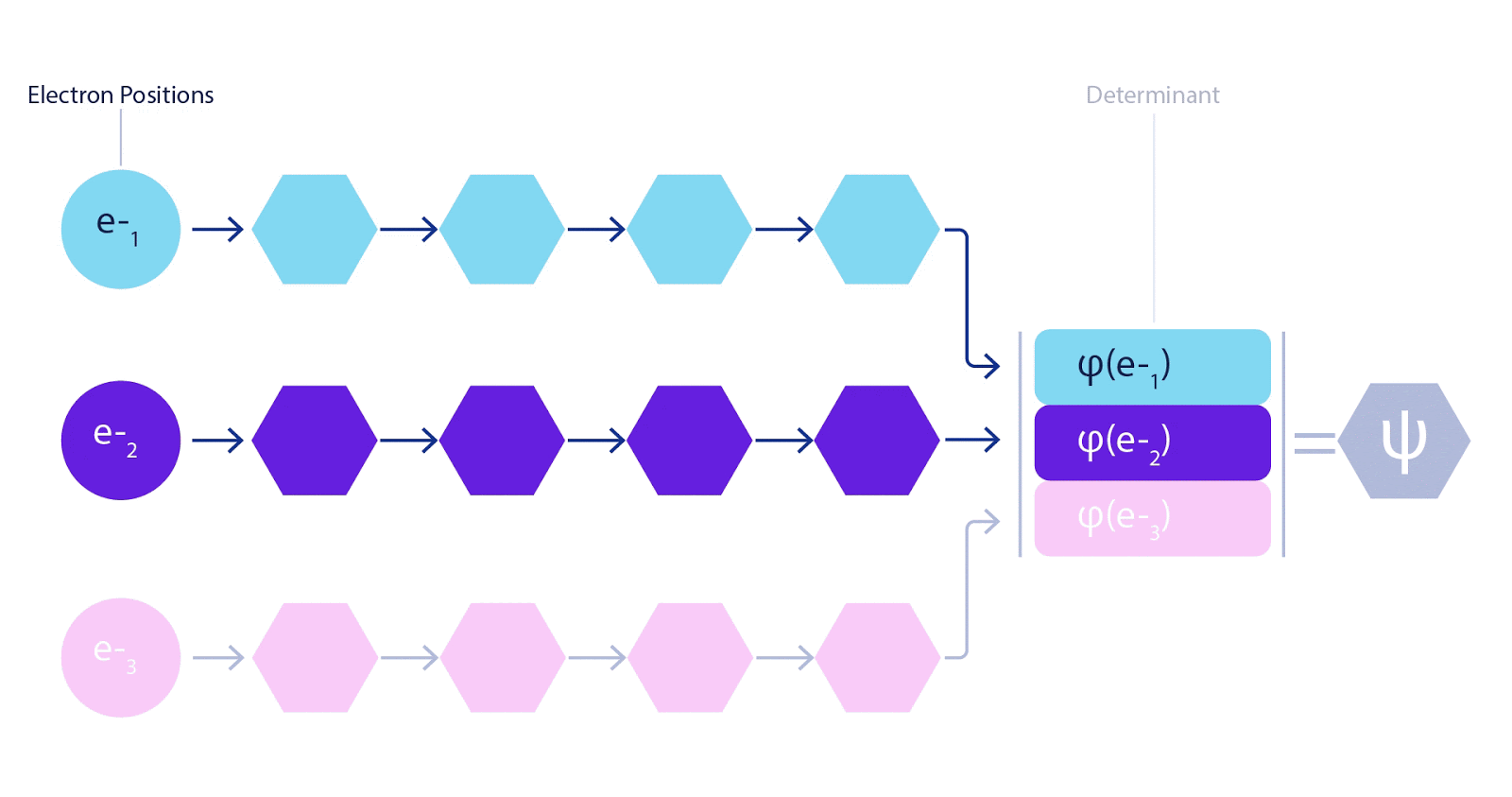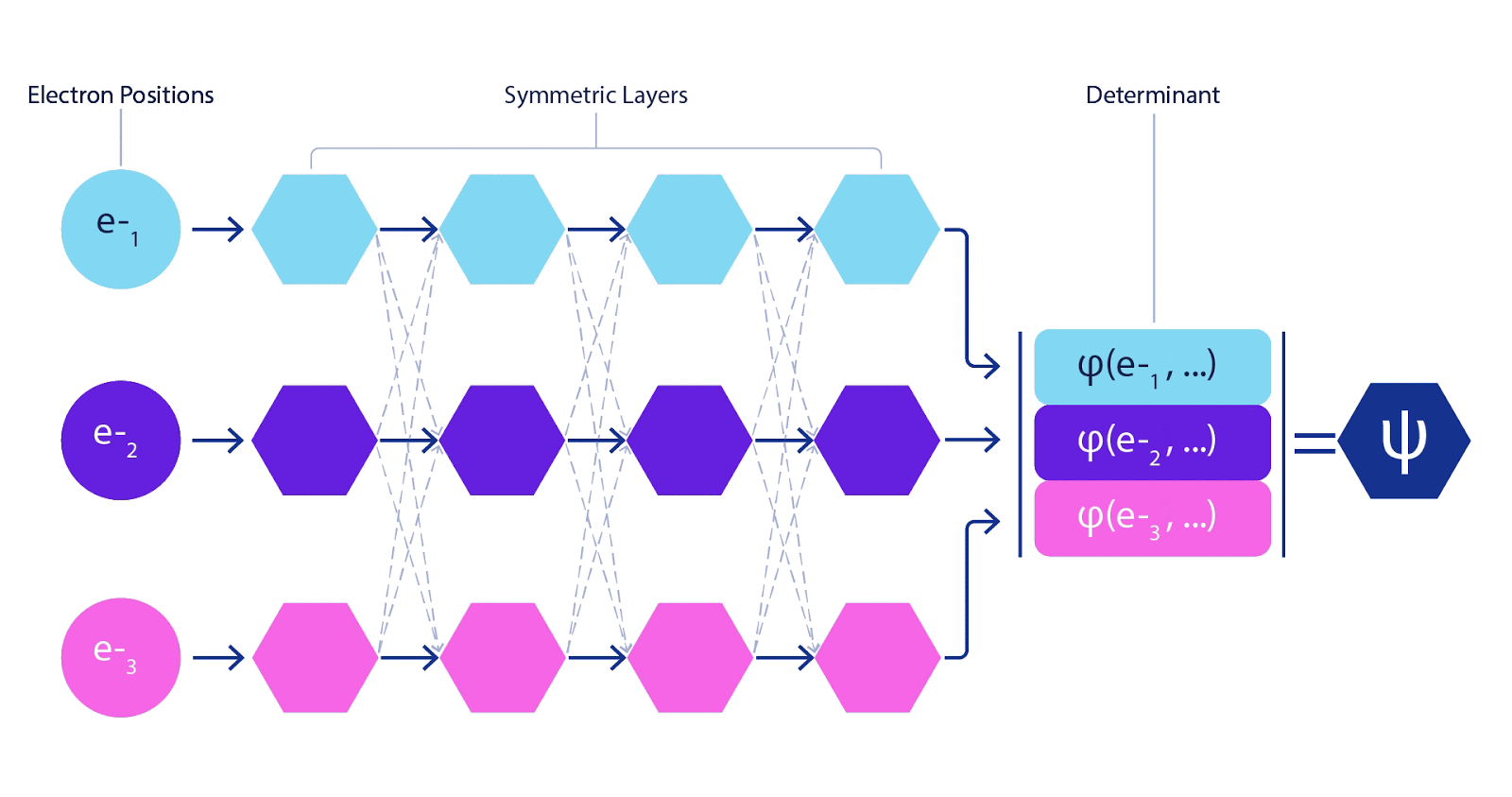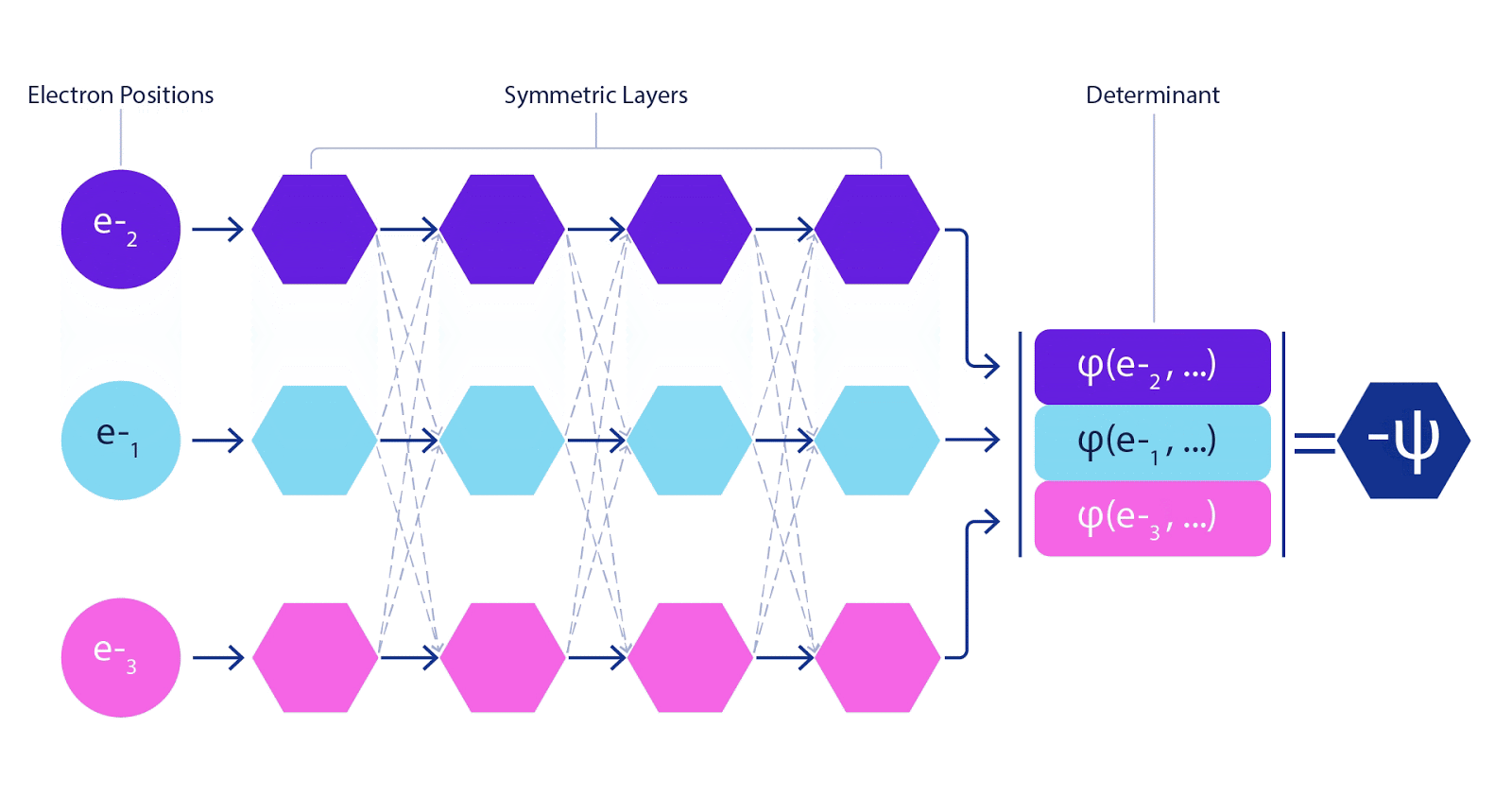
Deep neural networks can typically be much more environment friendly at representing advanced features than linear combos of foundation features. Within the FermiNet, that is achieved by making every operate going into the determinant a operate of all electrons (1). This goes far past strategies that simply use one- and two-electron features. The FermiNet has a separate stream of knowledge for every electron. With none interplay between these streams, the community could be no extra expressive than a traditional Slater determinant. To transcend this, we common collectively data from throughout all streams at every layer of the community, and cross this data to every stream on the subsequent layer. That means, these streams have the precise symmetry properties to create an antisymmetric operate. That is much like how graph neural networks mixture data at every layer. In contrast to the Slater determinants, FermiNets are universal function approximators, no less than within the restrict the place the neural community layers grow to be extensive sufficient. That signifies that, if we will practice these networks appropriately, they need to be capable to match the nearly-exact answer to the Schrödinger equation.
We match the FermiNet by minimising the power of the system. To do this precisely, we would want to judge the wavefunction in any respect doable configurations of electrons, so we’ve got to do it roughly as an alternative. We choose a random number of electron configurations, consider the power regionally at every association of electrons, add up the contributions from every association and minimise this as an alternative of the true power. This is called a Monte Carlo methodology, as a result of it’s a bit like a gambler rolling cube again and again. Whereas it’s approximate, if we have to make it extra correct we will at all times roll the cube once more. For the reason that wavefunction squared provides the likelihood of observing an association of particles in any location, it’s most handy to generate samples from the wavefunction itself – primarily, simulating the act of observing the particles. Whereas most neural networks are educated from some exterior information, in our case the inputs used to coach the neural community are generated by the neural community itself. It’s a bit like pulling your self up by your personal bootstraps, and it signifies that we don’t want any coaching information aside from the positions of the atomic nuclei that the electrons are dancing round. The fundamental concept, referred to as variational quantum Monte Carlo (or VMC for brief), has been round because the ‘60s, and it’s typically thought-about an inexpensive however not very correct means of computing the power of a system. By changing the straightforward wavefunctions based mostly on Slater determinants with the FermiNet, we’ve got dramatically elevated the accuracy of this strategy on each system we’ve checked out.
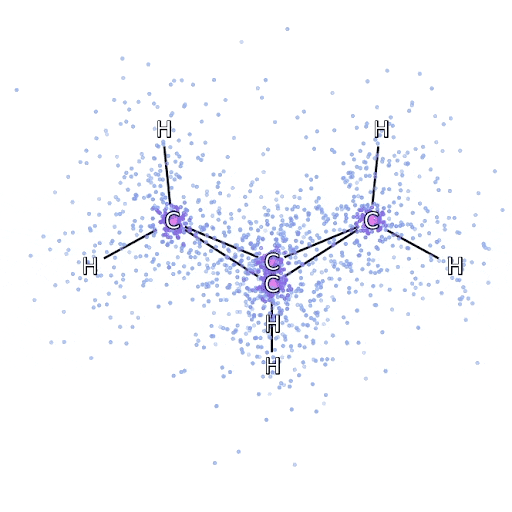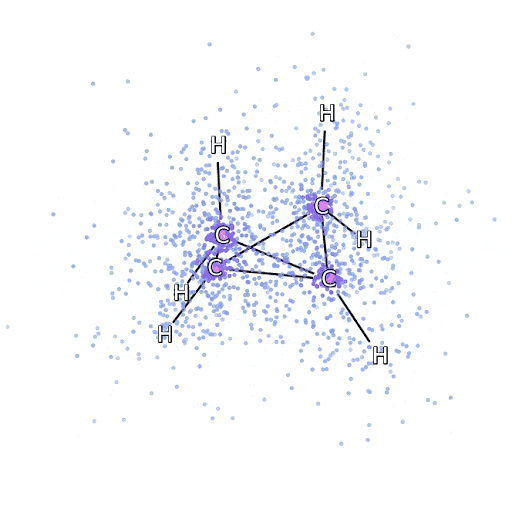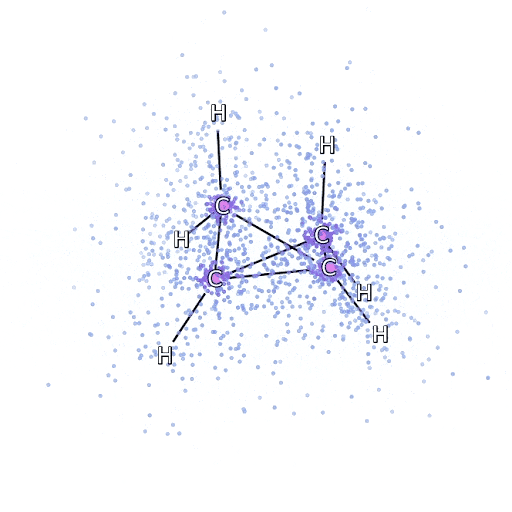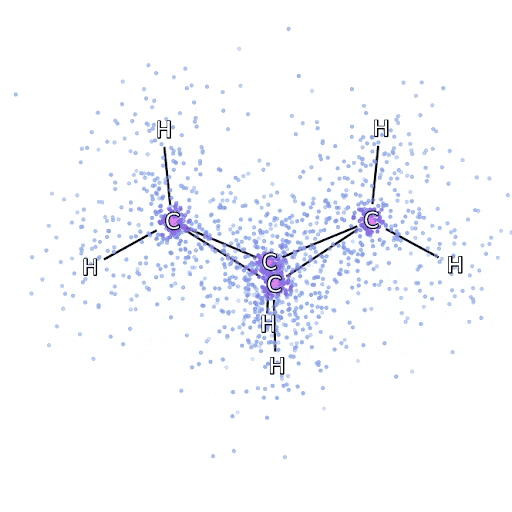
To ensure that the FermiNet actually does characterize an advance within the cutting-edge, we began by investigating easy, well-studied programs, like atoms within the first row of the periodic desk (hydrogen by neon). These are small programs – 10 electrons or fewer – and easy sufficient that they are often handled by essentially the most correct (however exponential scaling) strategies. The FermiNet outperforms comparable VMC calculations by a large margin – typically chopping the error relative to the exponentially-scaling calculations by half or extra. On bigger programs, the exponentially-scaling strategies grow to be intractable, so as an alternative we use the “coupled cluster” methodology as a baseline. This methodology works properly on molecules of their secure configuration, however struggles when bonds get stretched or damaged, which is crucial for understanding chemical reactions. Whereas it scales significantly better than exponentially, the actual coupled cluster methodology we used nonetheless scales because the variety of electrons raised to the seventh energy, so it might solely be used for medium-sized molecules. We utilized the FermiNet to progressively bigger molecules, beginning with lithium hydride and dealing our means as much as bicyclobutane, the biggest system we checked out, with 30 electrons. On the smallest molecules, the FermiNet captured an astounding 99.8% of the distinction between the coupled cluster power and the power you get from a single Slater determinant. On bicyclobutane, the FermiNet nonetheless captured 97% or extra of this correlation power – an enormous accomplishment for a supposedly “low cost however inaccurate” strategy.
Whereas coupled cluster strategies work properly for secure molecules, the true frontier in computational chemistry is in understanding how molecules stretch, twist and break. There, coupled cluster strategies typically battle, so we’ve got to match towards as many baselines as doable to ensure we get a constant reply. We checked out two benchmark stretched programs – the nitrogen molecule (N2) and the hydrogen chain with 10 atoms, (H10). Nitrogen is an particularly difficult molecular bond, as a result of every nitrogen atom contributes 3 electrons. The hydrogen chain, in the meantime, is of interest for understanding how electrons behave in materials, for example predicting whether or not or not a cloth will conduct electrical energy. On each programs, coupled cluster did properly at equilibrium, however had issues because the bonds had been stretched. Standard VMC calculations did poorly throughout the board. However the FermiNet was among the many finest strategies investigated, regardless of the bond size.
Conclusions
We predict the FermiNet is the beginning of nice issues to return for the fusion of deep studying and computational quantum chemistry. A lot of the programs we’ve checked out thus far are well-studied and well-understood. However simply as the primary good outcomes with deep studying in different fields led to a burst of follow-up work and speedy progress, we hope that the FermiNet will encourage numerous work on scaling up and lots of concepts for brand spanking new, even higher community architectures. Already, since we first put our work on arXiv final yr, other groups have shared their approaches to making use of deep studying to first-principles calculations on the many-electron downside. We’ve additionally simply scratched the floor of computational quantum physics, and stay up for making use of the FermiNet to powerful issues in materials science and condensed matter physics as properly. Principally, we hope that by releasing the supply code utilized in our experiments, we will encourage different researchers to construct on our work and check out new functions we haven’t even dreamed of.

